7200, 7200A, 7200CTS Detailed Feature Comparison
Unsure of which volatiles preconcentration system best fits your needs? This document can help you decide. All systems are certified for methods EPA-TO14a, TO-15, TO-15a, and China method HJ759.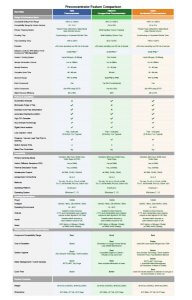
A New Cryogenless TO15 Canister Preconcentrator with Reduced System Carry-Over When Exposed to Higher Concentration Samples
Overview A new preconcentrator design is presented which eliminates the need for liquid nitrogen or complicated electronic cooling while performing US EPA Method TO15 analysis. A new technology called Multi-Capillary Column Trapping System (patent pending), or MCCTS, is used to concentrate all TO15 compounds at 35°C, which is achieved simply by using cooling fans. Two […]
7200CTS Cryogen-Free Preconcentrator Brochure
Entech is proud to release the world’s first multi-capillary column trapping system (MCCTS) patent pending, for the precise concentration of vapor phased volatile chemicals in the boiling point range of -50°C to >230°C without the need for liquid nitrogen or complicated electronic cooling. With over 28 years of continued improvements and industry feedback, the 7200CTS […]
Improved Performance And Dynamic Range For EPA Method TO-15 using the Entech 7200 and Agilent 7890B/5977 GCMS
Canister sampling and analysis for measurement of volatile chemicals is finding use in a growing number of diversified applications. This has required air laboratories to accommodate an even wider dynamic range of sample concentrations, and a growing list of compounds to include those once thought to be incompatible with canister sampling techniques.
Strong similarities between night-time deposition velocities of carbonyl sulphide and molecular hydrogen inferred from semi-continuous atmospheric observations
Strong similarities between night-time deposition velocities of carbonyl sulphide and molecular hydrogen inferred from semi-continuous atmospheric observations in Gif-sur-Yvette, Paris region.
7200 Operations Manual .NET
7200 Preconcentrator Operations ManualEntech 2023 Catalog
This is the complete Entech 2023 Catalog. Please note that not all sampling products are included. The Entech store is the best place to find photos and descriptions of every product we carry.
Cryogenic Pre-concentration Theory
A Three Stage Trapping Approach for EPA Method TO-15
7200 Preconcentrator
The 7200 takes the industry standard, 7100A’s 3-stage preconcentration and water management technology, and elevates it to a whole new level. Many of the past limitations often seen in rotary valve based devices are now eliminated by combining digital valve isolation control with advanced robotic autosamplers, dropping the potential for carryover and cross-contamination far below […]